欢迎您注册蒲公英
您需要 登录 才可以下载或查看,没有帐号?立即注册
x
导读
欧盟医疗器械法规(MDR)给全球医疗器械制造商带来了许多挑战。其中之一就是对将在欧盟内开展贸易的所有医疗器械制造商的标签要求产生重大影响。作为审核内容之一,标签不规范对于取得MDR证书和对于在欧洲销售医疗器械都有重大影响。 与 MDD 93/42/EEC 相比,欧盟 MDR 下的标签需要更多信息,因为设备安全性和临床有效性数据需要与用户(医务人员和患者/最终用户)透明共享)。欧盟 MDR附件 I 第 III 章“一般安全和性能要求”中涵盖了有关随医疗器械提供的信息的所有要求。 根据欧盟医疗器械法规 (MDR)(第 2 条)中的定义,“标签”是指出现在器械本身、每个单元的包装上或产品包装上的任何书面、印刷或图形信息。多个设备。贴标签过程的目的是识别医疗器械及其制造商,并传达有关安全、使用和性能的基本信息。它适用于医疗设备的用户,包括专业人士和消费者,以及相关第三方。

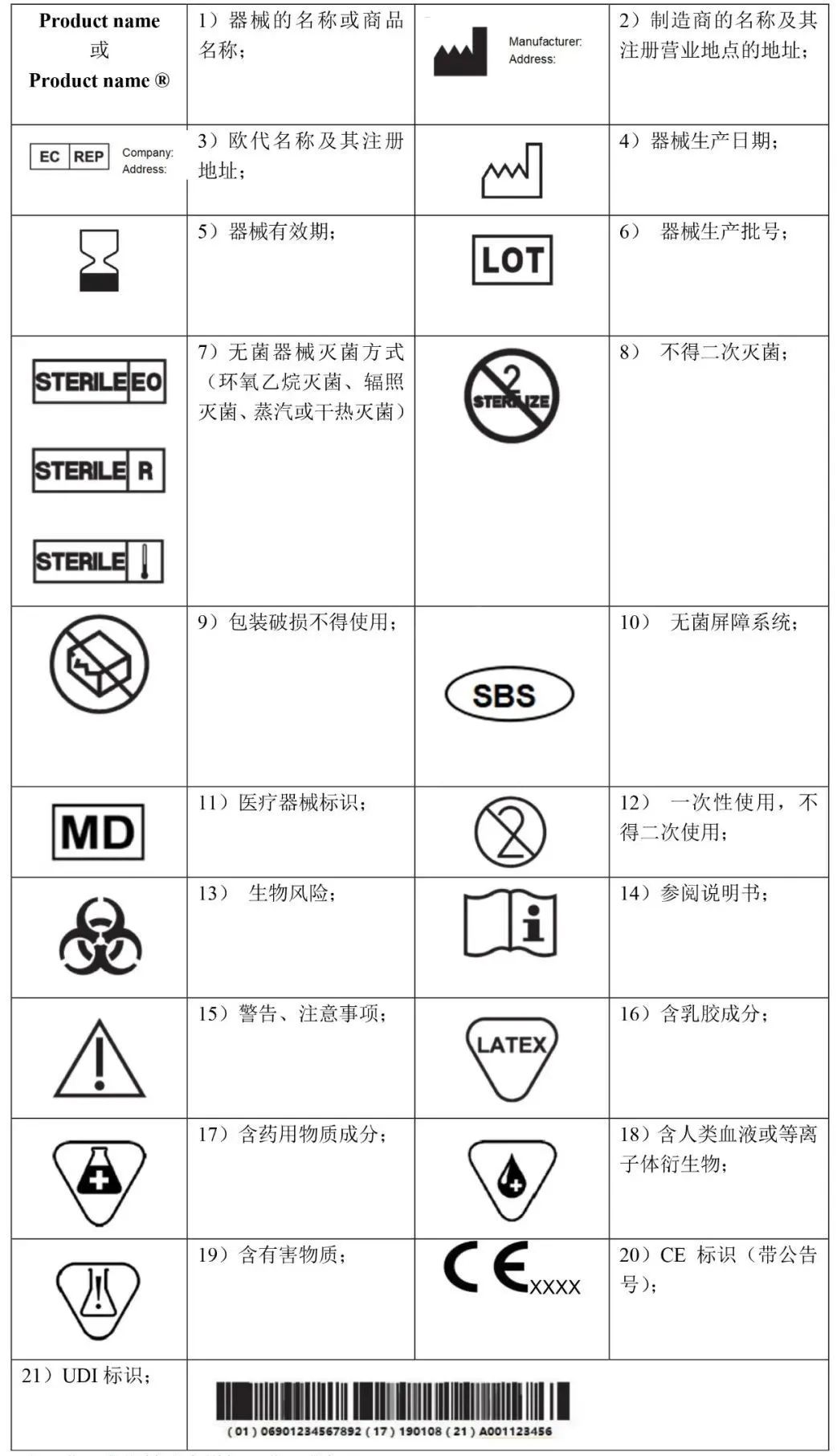
MDR 附录I中第III章23.2对于产品标签要求必须注明
(a) 器械的名称或商品名称; (b) 使用者识别器械所必需的详细信息、包装内容以及对于使用者不明显的器械预期用途; (c) 制造商的名称、注册商号或注册商标及其注册营业地点的地址; (d) 授权代表的姓名和授权代表的注册营业地点地址(若制造商在欧盟以外有其注册营业地点); (e) 若没有指明可安全使用的日期,则指明制造日期。若日期清晰可辨,制造日期可作为批号或序列号的一部分。 (f) 指明适用的任何特殊储存和/或处理条件; (g) 若以无菌方式提供器械,还应指示其无菌状态和灭菌方法; (h) 需要立即引起器械使用者和任何其他人的注意、需要采取的警戒或预防措施。 (i) 若器械用于一次性使用,则相应指明。制造商的一次性使用指示应在整个欧盟内保持一致; (j) UDI 载体应添加在该器械标签和所有更大包装上; (k) 标签应明显、清晰和不可磨灭地添加在器械或其无菌包装上。考虑到器械性质,无法或不适合将标签添加到器械上时,应将 CE 标识添加在包装上。CE 标识也应加贴在有使用明和任何销售包装中; (l) 应采用器械上市国(同时也是成员国)指定的欧盟官方语言编写,也可以采用预销往国的当地语言; (m) 标签上所需的信息应在器械本身上提供。若不可行或不适当,则某些或所有信息可显示在各单元的包装上和/或多个器械的包装上。
尝试遵守欧盟 MDR 标签要求时可能会遇到两个问题。一是确保涵盖所有必要的符号和信息。另一个是标签的大小。由于需要更多的符号和数据,最大的挑战将是如何将它们全部放在标签上。在标签设计过程中,请记住以下几点:标签和说明的媒介、格式、内容、易读性和位置必须与预期用户的技术知识、经验、教育或培训相匹配。此外,使用说明必须以预期用户易于理解的术语编写,并在适当的情况下补充附图和图表。 标签可以以人类可读的格式提供,并且可以用机器可读的信息来补充。
免责声明:本文为转发,如有侵权请尽快与我联系删除,我将会第一时间解决问题。
| 
 |手机版|蒲公英|ouryao|蒲公英
( 京ICP备14042168号-1 ) 增值电信业务经营许可证编号:京B2-20243455 互联网药品信息服务资格证书编号:(京)-非经营性-2024-0033
|手机版|蒲公英|ouryao|蒲公英
( 京ICP备14042168号-1 ) 增值电信业务经营许可证编号:京B2-20243455 互联网药品信息服务资格证书编号:(京)-非经营性-2024-0033





 发表于 2022-6-10 09:23:46
发表于 2022-6-10 09:23:46

 置顶卡
置顶卡 变色卡
变色卡