欢迎您注册蒲公英
您需要 登录 才可以下载或查看,没有帐号?立即注册
x
本帖最后由 Q821719617 于 2024-5-8 14:19 编辑
近年来,沙特阿拉伯已经在医疗保健方面投入大量资金并将继续增加支出,这使其成为医疗设备制造商感兴趣的市场。然而,想要在该国销售其设备的制造商首先必须满足监管要求,即他们必须在沙特阿拉伯获得其设备的授权。
沙特医疗器械上市合规流程
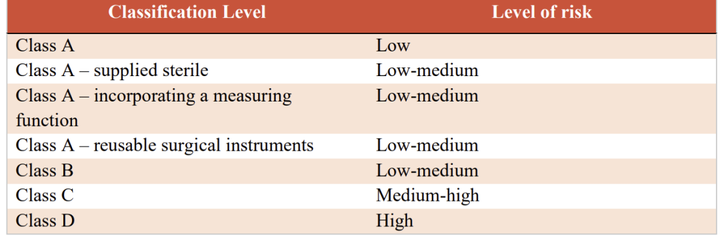
3.1 医疗器械风险分类 出口沙特医疗器械和体外诊断器械应依据MDS-G-008指南规定进行分类。
表1 医疗器械(不含IVD)分类
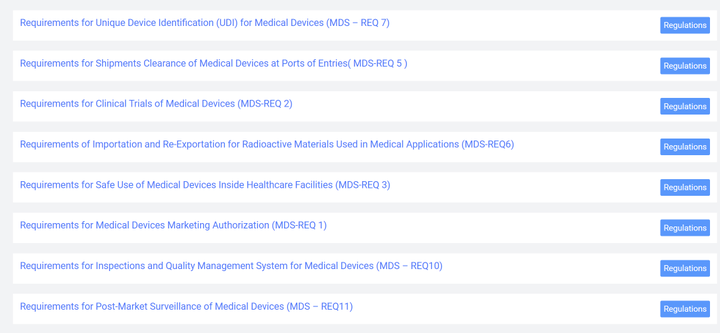
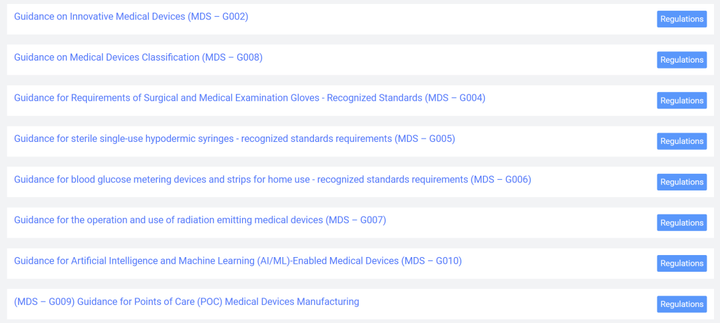
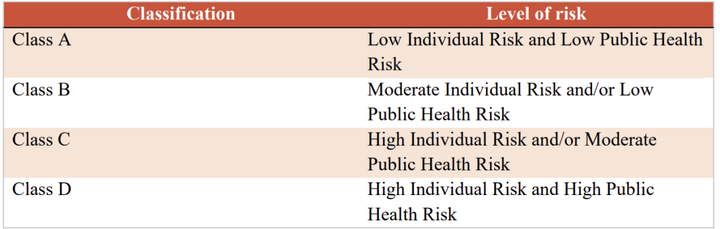
表2 体外诊断IVD分类 3.2 医疗器械上市合规流程 下图是沙特关于医疗器械的法规结构框架,从中可以看出制造商和授权代表需要完成MDMA评审程序,同时授权代表应完成MDNR注册。 作为中国制造商,应按照以下步骤完成器械上市前的合规程序。 1)指定授权代表 在沙特阿拉伯没有注册办事处的制造商需要在该国有授权代表 (AR)。AR必须在SFDA注册,然后才能提交医疗器械进行授权。 2)MDNR编号 SFDA负责维护在线医疗器械国家注册处 (MDNR)。该数据库列出了所有设备和位于沙特阿拉伯的公司。在设备投放市场之前,AR会提交相关信息并分配一个MDNR编号。 3)QM系统医疗器械制造商必须拥有符合 ISO 13485:2016标准的QM系统。相应的证书和指定机构的最新审计报告必须提交给国家食品药品监督管理局。 4)技术文件申报MDS-REQ 1 附件3和附件4分别列出了医疗器械产品和体外诊断器械的技术文件内容目录。技术文件的内容和格式基本参照STED的相关要求。 5)在线申报技术文件 6)支付申请和评审费用 7)当局评审,通过后颁发证书。 特别关注1:自2022年9月27日起,所有类别的医疗器械均应走MDMA(Medical Device Marketing Authorization)通道。不管是A类还是其他高风险类别的产品,均应符合MDMA的要求。海外制造商应委派当地授权代表,由其代表制造商进行MDMA注册。 特别关注2:SFDA已取消了MDMA GHTF申报路径,即原本医疗器械通过欧盟,美国,加拿大,澳大利亚,日本的认证或注册后便可完成审批的路径被取消。现在制造商只能通过MDMA TFA(Technical File Assessment)这一种路径获得许可。这一路径要求制造商准备并提交技术文件进行审核,对制造商的要求更为严格。 3.3 注册审批时间 MDMA审批时间为提交申请资料后2-3个月,更高风险等级的产品审批时间会更长
| 
 |手机版|蒲公英|ouryao|蒲公英
( 京ICP备14042168号-1 ) 增值电信业务经营许可证编号:京B2-20243455 互联网药品信息服务资格证书编号:(京)-非经营性-2024-0033
|手机版|蒲公英|ouryao|蒲公英
( 京ICP备14042168号-1 ) 增值电信业务经营许可证编号:京B2-20243455 互联网药品信息服务资格证书编号:(京)-非经营性-2024-0033





 发表于 2024-5-8 14:17:35
发表于 2024-5-8 14:17:35

 置顶卡
置顶卡 变色卡
变色卡