欢迎您注册蒲公英
您需要 登录 才可以下载或查看,没有帐号?立即注册
x
欧盟是中国药品出口最大的市场之一,很多本土药企期望能够提升自身综合实力,进入欧盟市场,但却对国内药品进入欧盟市场的条件和流程不熟悉。本期通过对话形式,让gempex德恩的GMP专家,为您解答国内药品进入欧盟市场的资质条件和流程,希望能够帮助本土药企走向国际市场。
问题1:国内药品进入欧盟市场有哪些条件? gempex专家解答:
在介绍国内药品进入欧盟市场的条件前,我们先了解两个概念:MAH和MIA。
MAH和MIA是国内药品进入欧盟市场的前提条件。对于国内的药品,如果你是MAH,那么我们需要寻找在欧盟境内取得MIA的公司。这里MIA的情况不同,进入市场的条件也会发生变化:
也就是说,凡是从欧盟境外进入欧盟境内的,需要通过有资质的进口商进入。
问题2:欧盟MAH和MIA有哪些资质要求? gempex专家解答:
1. MAH资质要求: 欧盟MAH的资质要求相对国内的MAH来说,会松一些。根据法规(EC)No.726/2004与指令2001/83/EC的法规要求节选如下:
MAH can be a natural person or a legal person but a resident in EU cannot be MAH the person must run a business and register in the government MAH of Centralized Procedure: established in EEA, have a permanent legal structure in EEA
意思是,欧盟MAH可以是自然人或者法人,包括任何人、企业、非盈利单位等。但自然人有以下要求:欧盟居民;需要有业务运营,并且进行官方注册。此外,如果是走集中审批制度*(见后)的MAH,那么其要求会更高一些,在EEA成立并且拥有永久在EEA内的合法组织架构。
2. MIA资质要求: MIA的资质要求有4点:体系要求(根据EU附录21的要求),人员要求(QP及管理投诉、退货、召回及供应链的人员),设施要求(有实际的进口场地 + QP的批发放场地)和取得证书。
System Requirement (EU Annex 21) Personnel Requirement (EU Annex 21): QP + personnel for complaints, returns, recalls, supply chain) Facility Requirements: Physical Importation site + QP batch Certification site Certification of MIA
如果在国内生产的药品(包括临床试验用药以及商业化产品)计划出口到欧盟市场,本土药企可以有多种选择,比如在欧盟寻找有条件的进口商,或者自己建立公司在欧洲做持有人和或者做进口商。
对于如何获得MAH及MIA资质,gempex德恩能够提供全方位的解决方案,包括法规策略咨询,体系搭建,资质申请协助等,如需了解,欢迎随时联系我们。
问题3:欧盟药品上市许可的申请流程是什么?
gempex专家解答:
欧盟药品上市许可常见的申请流程,概览如下:
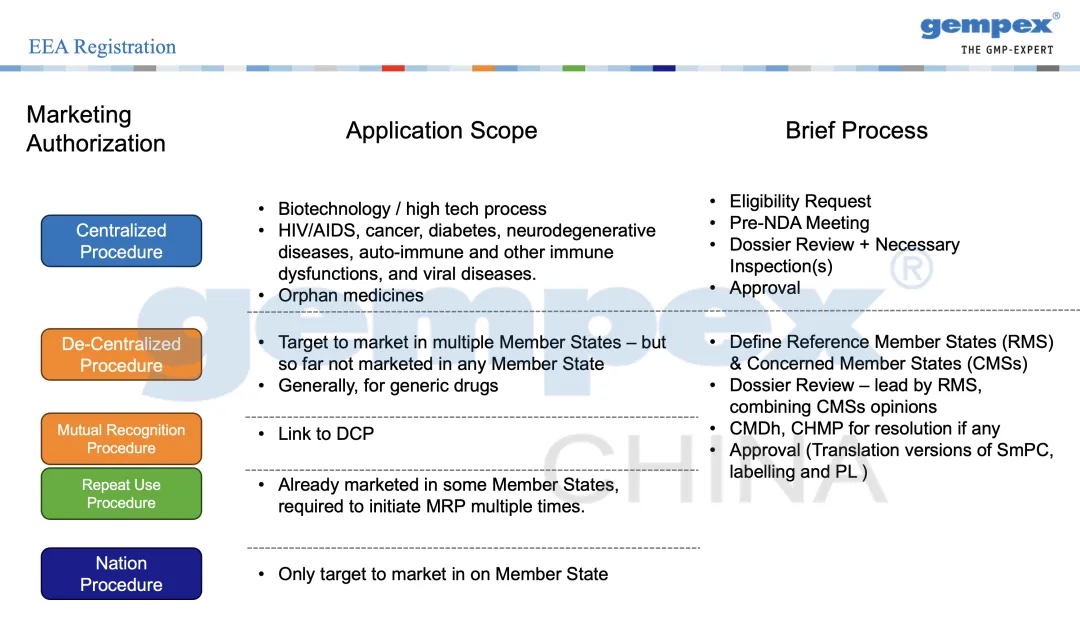
其中: 1. *集中审评流程(Centralized Procedure,CP): CP通过后可以在欧盟成员国以及三个EEA国家上市,但其对产品的范围有要求,主要适用于新药、复杂技术、特殊药品等。药企在递交申请的时候,EMA会对其资质进行评定是否属于范围内。如果是范围内,EMA官网会发布具体到每一步流程的清晰介绍,包括NDA前的会议、注册文件审评、相关的检查等,通过后批准。如果不属于EMA会推荐适用的流程。
2. 非集中审评制度或者分散审评制度(De-Centralized Procedure, DCP): 对于目标是在多个成员国上市销售药品的企业,且集中审评制度不适用的,可以选择非集中审评流程。非集中审评制度适用于尚未进入任何欧盟成员国的仿制药类产品,它会选择一个参考成员国(RMS)做主导,关联成员国(CMSs)共同审评提供审评意见。
3. 互认流程/重复使用流程(Mutual Recognition Procedure,MRP / Repeated Use Procedure, RUP): 互认流程(MRP)是指,产品已经得到欧盟成员国上市许可,可以通过该流程申请在其他成员国上市。MRP和DCP的区别在于有没有通过某成员国上市许可。一般而言,启动DCP流程的同时,其他关联成员国会自动启动MRP流程。
重复使用流程是指,产品已经在欧盟成员国上市(通过DCP或NP流程),希望扩展上市到其他成员国市场时,采用的流程与互认流程类似。
4. 单一成员国审评流程(Nation Procedure,NP): 如果产品只考虑进入某一个成员国,那么可以通过NP获得一个成员国的上市批准。将来需要拓展市场时,再通过MRP进入其他成员国。
每一种审评制度的适用范围、复杂程度以及要求有较大区别,在这里我们只给大家展示一下大体的内容,详情请参阅EMA/HMA以及各成员国官方的信息。
此外,药企在注册申请期间还会遇到各类检查,如QP检查、官方现场检查等,欲了解这些检查,可直接联系我们:020-2238 2526 或者通过邮箱 info-cn@gempex.com与我们联系。 今天专家的分享您都清楚了吗?
| 

 |手机版|蒲公英|ouryao|蒲公英
( 京ICP备14042168号-1 ) 增值电信业务经营许可证编号:京B2-20243455 互联网药品信息服务资格证书编号:(京)-非经营性-2024-0033
|手机版|蒲公英|ouryao|蒲公英
( 京ICP备14042168号-1 ) 增值电信业务经营许可证编号:京B2-20243455 互联网药品信息服务资格证书编号:(京)-非经营性-2024-0033






 发表于 2024-4-12 10:17:22
发表于 2024-4-12 10:17:22
 置顶卡
置顶卡 变色卡
变色卡